We explore the practical basics of managing an elevated ICP with impending herniation, including EVD troubleshooting, hyperosmolar therapies, and the role of craniectomy.
With Dr. Prem Kandiah, neurointensivist, transplant intensivist, and ammonia enthusiast, we explore the physiology of hyperammonemia, the nuances of its measurement and interpretation, and unpack some less-recognized causes, including infection by urease-producing organisms and malnutrition/gastric bypass.
Bernal, W. and J. Wendon, Acute liver failure. N Engl J Med, 2014. 370(12): p. 1170-1.
Bernal, W., et al., Lessons from look-back in acute liver failure? A single centre experience of 3300 patients. J Hepatol, 2013. 59(1): p. 74-80.
Kandiah PA, Nanchal R, Subramanian RM. Acute Liver Failure. In: Schmidt GA, Kress JP, Douglas IS. eds. Hall, Schmidt and Wood’s Principles of Critical Care, 5th Edition. McGraw Hill; 2023. Accessed August 09, 2023.
CRRT in ALF
Warrillow S, Fisher C, Tibballs H, et al. Continuous renal replacement therapy and its impact on hyperammonaemia in acute liver failure. Crit Care Resusc 2020;22:158-165.
Cardoso FS, Gottfried M, Tujios S, Olson JC, Karvellas CJ, Group USALFS. Continuous renal replacement therapy is associated with reduced serum ammonia levels and mortality in acute liver failure. Hepatology 2018;67:711-720.
Osmolar Shifts in ALF
Liotta EM, Romanova AL, Lizza BD, Rasmussen-Torvik LJ, Kim M, Francis B, et al.
Osmotic shifts, cerebral edema, and neurologic deterioration in severe hepatic encephalopathy. Crit Care Med. (2018) 46(2):280–9. doi: 10.1097/CCM.0000000000002831
Ammonia toxicity in Liver Failure
Dasarathy S, Mookerjee RP, Rackayova V, Rangroo Thrane V, Vairappan B, Ott P, et al. Ammonia toxicity: from head to toe? Metab Brain Dis. (2017) 32(2):529–38. doi: 10.1007/s11011-016-9938-3
Guo, R.M., et al., Brain MRI findings in acute hepatic encephalopathy in liver transplant recipients. Acta Neurol Belg, 2018. 118(2): p. 251-258
Kumar G, Taneja A, Kandiah PA. Brain and the Liver: Cerebral Edema, Hepatic Encephalopathy and Beyond. Hepatic Critical Care. 2017 Aug 7:83–103. doi: 10.1007/978-3-319-66432-3_8. PMCID: PMC7122599.
Ammonia in Cirrhosis
Gallego JJ, Ammonia and beyond – biomarkers of hepatic encephalopathy. Metab Brain Dis. 2025 Jan 15;40(1):100. doi: 10.1007/s11011-024-01512-7. PMID: 39812958; PMCID: PMC11735499.
Ammonia in in post bariatric surgery hyperammonemia
Kamel AY, Shah P, Pipek LZ, Shah A, Kandiah PA. Nutritional Emergencies. Current Surgery Reports. 2025;13(1):30. DOI10.1007/s40137-025-00465-9
Post Lung Transplant Hyperammonemia
Kamel, A.Y., et al., Hyperammonemia After Lung Transplantation: Systematic Review and a Mini Case Series. Transpl Int, 2022. 35: p. 10433.
Post cardiac arrest Hyperammonemia
Nojima T et al. Can Blood Ammonia Level, Prehospital Time, and Return of Spontaneous Circulation Predict Neurological Outcomes of Out-of-Hospital Cardiac Arrest Patients? A Nationwide, Retrospective Cohort Study. J Clin Med. 2022 May 4;11(9):2566. doi: 10.3390/jcm11092566. PMID: 35566692; PMCID: PMC9105173.
Ammonia Bowel Ischemia
Watari M et al. Ammonia determination as an early indicator in experimental superior mesenteric artery occlusion. Hiroshima J Med Sci. 1997 Dec;46(4):159-67. PMID: 9538566.
Study on undifferentiated causes of hyperammonemia
Maquet J et al. Clinical, biochemical, and molecular findings in adults with hyperammonemia: A French bi-centric retrospective study. Mol Genet Metab. 2025 Sep-Oct;146(1-2):109223. doi: 10.1016/j.ymgme.2025.109223. Epub 2025 Aug 13. PMID: 40834544.
Sakusic A, Features of Adult Hyperammonemia Not Due to Liver Failure in the ICU. Crit Care Med. 2018 Sep;46(9):e897-e903. doi: 10.1097/CCM.0000000000003278. PMID: 29985210; PMCID: PMC6095817.
Takeaway lessons
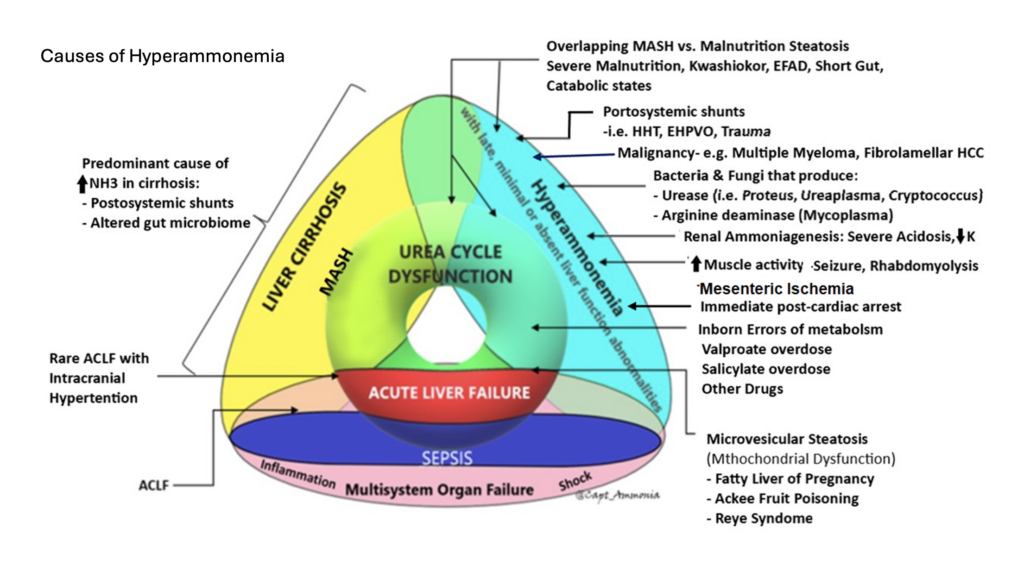
Effective ammonia clearance requires liver function, kidney function, gut function, and presence of skeletal muscles. It can also be compromised by A-V shunting bypassing the portal venous system (i.e. from enteric to systemic circulation; ask your radiologist to look for this as they may not otherwise report it).
Measuring the plasma ammonia level is not too technically difficult, but does have a time limit; allowing a sample to sit may falsely elevate the result. It’s therefore tough to do for outpatients. For inpatients, however, unless it gets forgotten on a table, it is generally not so difficult. Arterial vs venous doesn’t matter.
The peak ammonia on admission for cirrhotics is not as relevant as their cumulative ammonia burden over time (though of course this is not measurable). While difficult to interpret for those patients, a very high number is still very toxic, a very normal level probably absolves hepatic encephalopathy as a cause of altered mental status, and trending a number in between may help confirm response to therapy (eg the number should come down). A persistently elevated ammonia despite adequate catharsis may indicate need for a more aggressive regimen, or other attention to the underlying cause; a common cause would be a patient with GI bleeding, where persistent blood in the gut can cause persistent ammonia elevation.
Everyone agrees that ammonia should be followed in acute liver failure, where it correlates closely with risk of cerebral edema.
Checking ammonia in the patient without clear liver failure, but unexplained altered mental status, is reasonable—though an abnormal result will require a thoughtful approach to explaining it.
Cardiac arrest, especially with prolonged resuscitation, will reliably increase the ammonia level (if measured during or immediately after), as a direct effect of global ischemia.
Gut ischemia can elevate ammonia. Enterocytes require glutamine for energy; by interrupting this pathway, they may generate ammonia via anaerobic metabolism, much as other cells might generate lactate.
Medications, particularly valproate, can elevate ammonia.
Any elevated muscle activity or muscle breakdown, such as seizure or rhabdomyolysis, can generate nitrogen and hence transient hyperammonemia.
Any sarcopenia predisposes to hyperammonemia, as skeletal muscle is too sparse to reliably clear ammonia.
Inborn errors of metabolism involving the urease cycle are uncommon in adults but something to consider, especially if genetic testing is available to you.
Severe malnutrition, most often due to gastric bypass, can cause hyperammonemia. This is probably due to multifactorial causes, including sarcopenia and muscle catabolism, but malnutrition seems to induce a true hepatic steatosis is well (reversible if nutrition is restored). Radiographically and clinically this looks like MAFLD/MASH, but is not caused by metabolic syndrome or obesity, but the opposite state of malnutrition. (This phenomenon is also seen in Kwashiorkor.) Probably this is due to some synthetic dysfunction affecting beta oxidation and lipid metabolism, and/or absence of essential fatty acids—not clear.
Urease-producing bacteria can generate ammonia. This will usually be transient, since antibiotics will readily kill them. However, with a deep-seated infection such as an abscess, this may not be true and should be considered as an ongoing ammonia source. Examples include: Staph epidermidis and Staph saprophyticus, Helicobacter Pylori, Klebsiella, Nocardia, Cryptococcus, Pseudomonas spp., Corynebacterium, Proteus penneri, Providencia stuartii, and Morganella morganii
An especially important urease producer is ureaplasma, an atypical organism similar to mycoplasma, usually causing UTI or even STI. This will often not cause other signs of clinical infection, but can be a cause of ammonia production, and will not be grown on routine cultures (PCRs are needed on urine or BAL, or the Mayo Clinic has a blood PCR—all send-outs), nor covered with routine antimicrobials (atypical coverage, such as doxycycline, azithromycin, or levofloxacin is needed). This infection mostly occurs in the immunosuppressed, such as transplant patients, where it can cause occult pneumonia (an important and morbid cause of post-transplant hyperammonemia); consider it as well in anybody on rituximab or similar immunosuppression. It can also cause “sterile” joint effusions, so a patient on rituximab whose joint is tapped and grows inflammatory-but-aseptic fluid may be presumed to have progression of an underlying rheumatologic disease, have their immunosuppression escalated, and then end up with disseminated ureaplasma.
For the hyperammonemic due to malnutrition/gastric bypass: nourish with glucose and fats while limiting or holding protein completely in the acute period. This can be parenteral initially, as it’s easy to titrate protein in that fashion. Check and/or supplement everyone with micronutrients likely to be insufficient: high-dose thiamine, B6, L-carnitine, copper, zinc. Once ammonia stabilizes and clears, start to introduce protein cautiously, with the goal of eventually restoring an anabolic state to reverse the underlying steatosis. Go slow weaning everything to avoid rebounds.
Refractory hyperammonemia can be treated with dialysis (CRRT), though data is limited in this setting. Starting earlier may make sense, as osmolar clearance may be rapid once initiated and could precipitate cerebral edema if the osm load is already high. Some would add hypertonic saline with CRRT to try and mitigate this rapid osm drop, a common tactic in the ALF realm.
A common cause of proximal decompensation for these people is when micronutrient deficiencies starts to reduce gut motility, leading to poor tolerance of oral nutrition and anorexia. Once you fix these and the gut allows oral intake, their nutritional status can improve.
Lactulose and rifaximin can have some temporizing role in these patients, but a very short-acting one. They may also tend to worsen nutrition by accelerating gut transit time and filling the small-volume stomach.
The urease cycle scavengers sodium phenylacetate-sodium benzoate may help accelerate clearance of ammonia, if renal function is intact (they convert ammonia to PAGN which can be renally cleared). However, they are not a magic solution, take time to work, and do not address the underlying problem.
We dive into the common dilemma of when to give stress-dose corticosteroids in septic shock, with Dr. David Janz, pulm/crit intensivist with a Masters of Science in Clinical Investigation, former director of the Clinical Research Unit for the critical care section at LSU, founding member of the Pragmatic Critical Care Research Group, and associate Chief Medical Officer for LCMC.
The purported effect of corticosteroids in septic shock is to increase sensitivity of peripheral catecholamine receptors, either to endogenous or exogenous adrenergic hormones.
In the four large RCTs of this topic, two showed a beneficial outcome effect and two did not. The two positive trials also added fludrocortisone to the backbone steroid (usually hydrocortisone), though it seems dubious that this was the key difference (fludro has also been studied separately without effect). Those trials also generally enrolled sicker patients, i.e. on higher doses of pressor and more organ failure.
It is probably reasonable to add steroids when on escalating doses of your first-line pressor (e.g. norepinephrine), or when thinking of adding vasopressin, though the SCCM/Surviving Sepsis guidelines now suggest just giving steroids to everyone in septic shock, presumably to simplify decision-making, particularly for non-experts.
Most trials have used hydrocortisone 50 mg q6h or thereabouts, or an equivalent dose as a continuous infusion. The latter makes some physiologic sense but has not been shown to be beneficial head-to-head trials. Most likely, the exact dosing strategy is not too important. Alternate steroids are probably also reasonable, i.e. any moderate-dose parenteral corticosteroid probably gets you the desired effect.
Many trials continued steroids until the patient left the ICU, even if pressors have been weaned off. Most clinicians would probably stop them sooner than this, although we should acknowledge that their effect in raising blood pressure is reliable (sometimes helping to discontinue pressors as much as several days sooner), so continuing them longer could conceivably help with disposition (i.e. help them leave and stay out of the ICU), which is a good thing for hospitals and probably patients.
Dr. Janz stops steroids when the pressors are stopped, with no taper or wean. Some would do a short taper, but the more complex you make the process in a busy ICU, the more likely that someone will forget to discontinue them altogether, and that the patient will stay on steroids for their whole hospitalization (or forever). There is no physiologic suppression of the native steroid production with these durations of steroid therapy (usually 3-4 days, almost always <1 week), and an abrupt halt is what was done in the trials.
Etomidate has been repeatedly shown to cause a measurable decrease in cortisol levels, even after single doses (e.g. for intubation). However, this has not been shown to be associated with any negative outcome (most recently in the EVK trial).
There is very little data on whether steroids should be used in shock from other etiologies. It may make some sense in other inflammatory states, such as pancreatitis, post-CPB vasoplegia, etc. But we really don’t know. In a mixed shock state where sepsis is present along with something else like cardiogenic shock, Dr. Janz uses steroids; otherwise no.
Whether to always augment the home steroid dose in chronic steroid users, even without shock, has shifted—in the past, endocrinology guidelines were to always do this empirically (e.g. in steroid users undergoing surgery or some other stressor), now they still generally favor this but are moving towards increasing the home dose only if signs of adrenal crisis develop.
We explore the fascinating intricacies and unique features of the burned critically ill patient, with Clint Leonard, NP in the burn ICU at Vanderbilt and ABLS instructor.
We discuss the evidence and practicalities of spontaneous breathing trials and ventilator weaning with Martin Tobin, pulmonologist and intensivist, author of seven textbooks including “Principles and Practice of Mechanical Ventilation,” researcher, and pioneer of the entire concept of SBT as well as the RSBI (or “Tobin index”).
The common practice of pressure support trials on 5/5 cm H2O is unwise, because it’s too easy; even these “modest” amounts of support are much more than they will have after extubation, decreasing work of breathing by ~40%.
The idea that pressure support helps negate the resistance of the endotracheal tube is also erroneous; in the typical patient with a typical airway, the diameter of the native airway after extubation, and hence the work of breathing, will roughly approximate that of breathing through the ET tube (due to edema, etc). In other words, don’t try to negate that resistance; it will still be present later.
For Dr. Tobin, the sensible alternatives are T-piece or blow-by oxygen, or a 0/0 trial. He likes T-piece as it’s highly controllable for research, but those who prefer the patient to be attached to the ventilator can certainly do 0/0. (Note that no modern ventilator actually drops to zero pressure, usually offering about 1 cmH2O of support, but this is negligible.)
Each additional day on the ventilator increases the risk of VAP by about 1% and VAP has a mortality of about 70%. Getting off the vent ASAP is a vital goal.
Dr. Tobin thinks the idea of physiologic PEEP occurring in patients breathing on their own, including the obese, is a fallacy; once extubated, no PEEP is present.
There is no advantage in an SBT beyond 30 minutes; 30 will answer the question.
Respiratory muscle fatigue occurs in the vast majority of ICU patients, including after SBT; it has not been shown in fatigue studies using twitch stimulation of the phrenic nerve and similar work. If it does occur, it takes about 24 hours to recover.
The best marker of success on SBT is probably using esophageal manometry to assess work of breathing using the transpulmonary pressure. Short of this, look for clinical findings of work of breathing by visualization and palpation: any phasic contraction in the sternomastoids, phasic movement in the thyroid cartilage, and phasic recession in the intercostals/suprasternal fossa/supraclavicular spaces. Oxygenation is obvious from oximetry, and ventilation only needs to be queried if you have a concern about hypercarbia.
There is no role for the RSBI in evaluating SBT success. This tool was derived by Dr. Tobin as a screening tool for SBT readiness, i.e. a protocolized way to push clinicians to perform SBT earlier than they otherwise might by judgment alone. Patients should be screened by briefly placing them on 0/0 or T-piece, waiting about 1 minute to normalize breathing, then monitoring the RSBI for 1 minute. If <105, SBT can probably be done safely and has a good chance of success.
Transient apnea after transitioning to a spontaneous mode is usually not due to drugs, but stimulation of the Golgi tendon organs and mechanoreceptors caused by mechanical ventilation; this takes some time to reset.
The first treatment for a patient who appears agitated during an SAT/SBT is to speak to the patient, reassure them, and bring a level of calm. Dr. Tobin is not a fan of SBT with continued sedation as he would rather not muck with the medulla oblongata in these sensitive patients.
Dr. Tobin is not a huge fan of planned extubation to NIPPV except in niche cases; he would rather wait and see them failing, given the discomfort and risks associated with using CPAP/BiPAP. (Extubation to HFNC is no problem.)
In general, the SpO2 should be no higher than 90%. This places the patient on the steep portion of the oxyhemoglobin dissociation curve, making a fall in oxygenation immediate and obvious; a higher goal blunts the utility of this diagnostic tool.
For chronic vent weaning with a tracheostomy, trach collar trials are superior to a weaning pressure support strategy.
A protocolized approach to weaning is the only way to expedite the process; relying on clinician judgment and gestalt will always result in longer vent times.
We explore the vagaries and nitty-gritty of drugs for seizure termination, including benzos and ASMs, with the great Tom Bleck, MD MCCM FNCS, neurointensivist, professor, and founding member of the Neurocritical Care Society.
In the RAMPART study, IM midazolam (10 mg) achieved faster seizure control than IV lorazepam (4 mg), with most of the difference accounted for by the time needed to start the line. So if there’s no IV, use IM midazolam.
If there is an IV, there is no data on superiority of any benzo over any other. This makes it appealing to use lorazepam, simply because we have the most data surrounding its use. But use whatever.
It is often hard to truly know the timeline since seizures began, other than those already on EEG monitoring. Probably in many published populations, benzo non-responders may have been patients who were already seizing for some time. As soon as seizures begin, benzo-sensitive gaba A receptors immediately begin to be replaced by benzo-insensitive receptors (a phenomenon not seen when benzos are used for other applications).
Most seizures, even in the ICU, end in about 90 seconds. If you don’t witness the onset, you should probably plan to treat it. If you do, you should prepare to treat, but wait five minutes to see if it stops, our current cutoff for status epilepticus—most will stop before then. The caveat is that it’s not impossible some of these people who appear to stop convulsing have simply become non-convulsive… this generally takes longer (30+ minutes), but in critically ill patients all bets are off.
Meaningful respiratory dysfunction from benzos for seizures is vanishingly rare. Most patients who get intubated due to benzo-related sedation do so out of clinician preference, not true need. Consider a side-lying rescue position to maintain the airway, or an oral or nasal airway.
The VA Cooperative trial used a lorazepam dose of 0.1 mg/kg, one time, no cap. Most of the current guidelines now suggest giving a 4 mg dose, then repeating if necessary. If this still leaves you under 0.1 mg/kg, a third dose to finish it out is reasonable. Giving more than 0.1 mg/kg is probably not more effective. Six minutes between doses is sometimes recommended, but most would go closer to 4-5 minutes at most.
The goal is cessation of convulsions, or of any other clinical signs of seizure (twitches, eye deviation, etc). Ideally, the patient would then wake up promptly, but this is a rarity, especially in the ICU. This then leaves the question of whether the patient is still seizing, merely non-convulsively. This needs an EEG to answer, but the risk to the brain once convulsions cease is probably less, so you can probably wait for the EEG (rather than giving more empiric doses of benzos).
If not rousing, however, you should probably treat with an anti-seizure medication, unless rapid access to EEG can prove lack of ongoing seizure. The three studied in the ESETT trial were: levetiracetam 60 mg/kg, valproate at 40 mg/kg, or fosphenytoin at 20 mg/kg, all of which had about 40% response rate. This was used in patients still convulsing at the time, but efficacy can probably be extrapolated to non-convulsive patients (or prevention of the next seizure in delicate patients). The study was not powered to look at subgroups, and all three seemed equally effective.
Levetiracetam is most popular by dint of its ease of use, especially now that it’s available by IV push, and there are very few times it would be the wrong choice in this context. (Even in renal failure, the same load can be used; maintenance will simply be lower/less frequent.) For long-term use, it does sometimes cause behavioral problems, but that is not an acute issue.
60 mg/kg is probably the right dose in most patients. Possibly less is effective in some cases, but it doesn’t seem to have toxicity at this dose, and higher doses probably reach adequate brain levels faster.
When to intubate and induce general anethesia? For Tom, only if they’re still convulsing (after benzo + ASM), or proven to still be seizing by EEG. But it’s an open question. Rapid EEG devices are a great tool here. If there is absolutely no access to EEG, you should consider transfer to a center that has it, probably within an hour or so if not waking.
There may be some role for inducing “procedural sedation” with propofol without intubating, which will terminate seizures in some cases. But this is a challenge for most non-anesthesiologists.
Most induction meds for intubation have some anti-seizure effect, including etomidate, so there’s not much reason to use an unusual drug here. However, do use some kind of sedative, even if they’re already obtunded (please don’t merely paralyze).
If paralyzing with rocuronium (not a bad idea, due to the risk of hyperkalemia in some of these patients), consider reversing it after, if you plan to use clinical markers to monitor their status.
Once intubated, high dose benzo (usually midazolam) or propofol? No data on this, but benzos tend to cause more problems, such as long weans due to metabolites. Most ICU folks are more comfortable with high dose propofol.
If using midaz, use a loading dose of 0.2 mg/kg (loading propofol is probably not necessary though it was done in some trials), then start an infusion of 0.2 mg/kg/hr. As tachyphylaxis sets in fast, you may need to frequently increase the dose afterwards (hourly or so), tough to do without an EEG; propofol may be a better choice without continuous EEG.
Tom would empirically use about 80 mcg/kg/min propofol if hemodynamically tolerated, if EEG was unavailable.
Ketamine as an adjunct to general anesthesia has useful hemodynamic properties (e.g. added to propofol to combat hypotension), but also seems to have an effective anti-seizure effect on its own. It’s less clear about using it as monotherapy. For effective seizure therapy, go for a completely dissociative dose (e.g. 5 mg/kg).
Not all epileptiform activity on EEG needs to be treated by escalating therapy. This is pertinent to the rapid EEG devices that try to report seizure via algorithm, as they’ll generally alert for anything seizure-like, whereas not all of these truly need treatment.
Usually with propofol, barbituates, or inhalational anesthesia, the initial goal is burst suppression with 10 second suppression between bursts. (Even this may only control about 1/3 of patients, who may need complete flatline.) If using midazolam or ketamine, however, you may never achieve burst suppression (only around 1/5 ever will), yet you may still achieve seizure control.
Frequency of monitoring of continuous EEG depends somewhat on the situation, eg is the patient still trying to achieve seizure control/suppression or have they reached a fairly steady state. Note: an occasional self-terminating seizure (e.g. one a day) may not mean a patient is failing weaning, as an occasional seizure in the outpatient setting is not exactly an emergency.
Levetiracetam has been dosed as high as 6 g per day, which is usually tolerated, although behavioral problems are common. Adding additional agents is usually better than megadoses, but Tom prefers to add them one by one if seizures prove refractory; adding multiple agents at one makes it hard to pinpoint the cause of drug reactions. Clobazam is favored by some to help wean anesthesia.
Tom would most often use valproate as a second line. A single dose is safe even in pregnant women (pregnancy was not screened in ESETT, though it wouldn’t be continued in known pregnancy). Fosphenytoin is still reasonable, though has more complications, and was the most associated with intubation in ESETT.
Lacosamide is often underdosed; it’s best loaded with 10-13 mg/kg, monitoring the PR interval.
Phenobarbital is a good drug, especially in difficult to control patients, but start with an aggressive general anesthetic dose: 20 mg/kg load, usually hitting a serum level ~20 (then taper), and may need to go to 100-150 mg/kg to allow weaning of general anesthesia. Some patients will even wake up on these doses once they adapt. Halflife is around 120 hours, so if you make a change, give a load or expect to wait for steady state; pentobarbital is faster, but its cost has been exorbitantly raised, so if you use it, maybe load, then switch to phenobarb.
In new onset refractory status (NORSE), adding therapies fairly early like anakinra or tocilizumab (as they often have ample IL-1 or IL-6) can be wise. Treat vitamin deficiencies; vit D deficiency may be a marker that supplementation is beneficial. A ketogenic die can be tried fairly early, though it’s a lot of work for the dieticians and pharmacists to eliminate carbohydrates from meds (eg dextrose); the target beta-hydroxybutyrate level is not clear. Ganaxolone probably has little role now.
In general, partial/focal seizures probably do not need to be treated as aggressively as generalized seizures. There is likely a spectrum, based heavily on the extensiveness of seizure; complex partial seizures (focal with altered awareness) that persist past 10 minutes or so should probably be treated. Most would not intubate/induce general anesthesia for these, but would treat with ASMs, and benzos may not be needed. Absence status seems to cause no damage regardless of duration, though they can be terminated by benzos, and ethosuximide can be used.
If focal seizures arise from a foci such as intracranial hemorrhage, it’s usually worth using an ASM to try and prevent generalization, but understand that sometimes the focal seizures can’t be suppressed. (And if there’s a foci like a tumor, remove it—it will help.) Vagal nerve stimulators or direct brain stimulation may help with focal seizures too, and maybe transcranial current stimulation, all areas of research.
Don’t despair. About 35% of patients with super refractory status will return to their baseline. (35% will also die, but that’s not bad odds.)
We dive into fluid resuscitation in sepsis, with Dr. Jon-Emile Kenny, pulmonary and critical care physician, author of the physiology textbook Heart-lung.org, and inventor of the FloPatch device.
Disclosures: Dr. Kenny appears here as both a clinician as well as a representative of his company and product, and should be presumed to retain a degree of bias in this discussion. However, his appearance is not part of a commercial relationship with our show; no compensation was provided, and neither he nor his company have any input in the episode’s content, nor the right to review it (or prevent its publication) after recording.
Before giving IV fluids, ask: 1) Is there an indication to give fluids? 2) Is giving fluids safe? 3) Will giving fluids be effective?
One of the most common misconceptions is that safety and efficacy are opposite ends of a spectrum, and efficacy necessarily implies safety. This is not so. Think instead of fluid like a drug, which could be effective, yet also dangerous (e.g. anaphylaxis), or vice versa. A volume overloaded patient could be fluid responsive, but giving fluid could be a poor idea.
Interpreting heart function and cardiac output is difficult in sepsis, particularly on the venous or filling side. Many have some degree of diastolic dysfunction, which sepsis itself can induce.
Kenny likes to phenotype patients into a 4-quadrant grid, similar to the traditional Diamond-Forrester heart failure classification, characterizing a patient as either wet/dry and normal/low cardiac output. POCUS can be used to assess both of these; LVOT VTI >17-20 is a normal-ish stroke volume and IVC is a surrogate for preload.
The only phenotype likely to benefit from fluid is the cold, dry patient (warm patients don’t need fluid, wet patients are unlikely to respond and maybe shouldn’t have it even if they will), although in sepsis, 20-30% of even this group are not fluid responsive and fluid will simply congest them.
Using BP response to fluid challenge is insensitive; in a significant number of patients, cardiac output will increase but BP will not. A marker of flow, e.g. doppler ultrasound, is more sensitive.
The FloPatch is a wireless, wearable, continuous-wave doppler ultrasound. It adheres over the neck and continuously monitors both the carotid and jugular vessels. The jugular provides a CVP-like waveform for qualitative clinician inspection, while the carotid is used to automatically measure the systolic flowtime duration, which is associated with stroke volume (better evidence than calculating a stroke volume).
The height of the carotid waveform may change with alterations in inotropy, afterload, and other factors, but if those are consistent, fluid responsiveness is best associated with the duration alone (base of the triangle).
An increase in flowtime duration of >7 milliseconds after a preload challenge (Trendelenburg position or ~250-300 ml rapid fluid bolus) is associated with 10% increase in stroke volume. (This cutoff is meant to match existing literature on fluid responsiveness.)
We discuss propofol infusion syndrome (PRIS) and propylene glycol toxicity from lorazepam infusions, with medical toxicologist Dr. Jerry Snow, director of the toxicology fellowship at Banner University Medical Center in Phoenix.
PRIS is a defect in the electron transport chain leading to a failure of ATP production and fatty acid metabolism. There seems to be a susceptibility in some part of the population, but not a clearly understood or monofactorial one. People with mitochondrial disorders are at higher risk, but there is no definitive testing for PRIS risk.
PRIS has mostly been described with infusions greater than ~67 mcg/kg/min infusions, running for >48 hours.
The most common presentation is some combination of: elevated lactate, rhabdomyolysis, and new cardiac changes (which may be varied, including new bundle branch block, bradycardia, Brugada-like ST elevations, and changes in function on echo). Trending lactate and CK periodically on high-dose propofol is not a bad idea.
Triglyceride elevation probably has some association with PRIS, as it is also associated with high propofol doses, but there is not a direct link.
The primary treatment is stopping propofol and supportive care. There have been some case reports of ECMO being used.
It is not clear whether patients might be treated by dose-lowering propofol rather than stopping entirely, but it would be a fairly bold move; a safer option might be discontinuation and later rechallenge, but many experts recommend avoiding propofol in the future. Data is limited, but it should probably be added to allergy lists.
Propylene glycol is a toxic alcohol used as diluent for lorazepam, diazepam, and phenobarbital infusions. It is lower amounts in the latter two, and they are less often used, so toxicity is almost always in lorazepam, and almost always in infusions (not intermittent boluses).
It is associated with higher infusion rates for prolonged periods. This is probably above >6-7 mg/hr, or >0.1 mg/kg/hr, or >1mg/kg/day, depending on who you ask.
Propylene glycol is an alcohol, which behaves similarly to other toxic alcohols like ethylene glycol; it creates an elevated osmolar gap, and is metabolized via alcohol dehydrogenase (ADH) to lactate, creating a lactic acidosis.
Presentation is predominantly an unexplained lactic acidosis. An elevated osmolar gap will help confirm. Mental status can be affected as well. Trending a daily lactate and/or osmolality is not a bad idea on high-dose lorazepam infusions.
There is no common confirmatory testing, although some centers can probably obtain propylene glycol levels.
Treatment of propylene glycol toxicity is predominantly stopping or weaning the drip and supportive care. In the most severe cases, it can be treated similarly to other toxic alcohols, including fomepizole and/or renal replacement therapy (especially in patients with renal failure who are more likely to accumulate the compound and its metabolites). It probably does not need to be listed as an allergy/drug reaction.
Brandon and Bryan mock up a goals of care discussion for a critically ill patient, and reflect on the right and wrong ways to execute this complex procedure.